|
Nr.
|
Frage
|
Antwort
|
|
1
|
Was bedeutet MDR?
|
"MDR" heißt Medical Device Regulation.
Die neue EU-Medizinprodukte-Verordnung (Medical Device Regulation – kurz: MDR) ist am 25. Mai 2017 gemeinsam mit der ebenfalls neuen Verordnung über In-vitro-Diagnostika (IVDR) in Kraft getreten.
|
|
2
|
Was passiert mit der bisherigen europäischen Richtlinie MDD (Medical Device Directive)?
|
Die neue EU-Medizinprodukteverordnung (MDR) ersetzt die aktuelle Medizinprodukterichtlinie (93/42/EWG) sowie die Richtlinie über aktive implantierbare medizinische Geräte (90/385/EWG).
|
|
3
|
Was passiert mit dem österreichischen MPG (Medizinproduktegesetz)?
|
Das bisherige Medizinproduktegesetz (MPG) wird durch ein neues Medizinproduktegesetz (MDR) abgelöst. Es regelt z. B. Anforderungen an Medizinprodukteberater, neue Zuständigkeiten für Aufsichtsbehörden und nationale Aspekte der klinischen Prüfung.
|
|
4
|
Warum wurden die bisherigen Medizinprodukte-Richtlinien abgelöst?
|
Einer der Gründe, die bisherigen Richtlinien abzulösen, ergab sich durch den sogenannten PIP-Skandal. Ein Hersteller verwendete in krimineller Absicht für Brustimplantate Industrie-Silikon anstelle hochreinen medizinischen Silikons.
Nach Bekanntwerden dieser Vorgänge ergaben sich Überlegungen, wie man zukünftig derartige kriminelle Prozesse durch eine Verschärfung der Anforderungen an die Zulassung und damit an das Inverkehrbringen sowie an die Marktüberwachung verhindern könne.
|
|
5
|
Was ist der Unterschied zum bisherigen MPG (Medizinproduktegesetz)?
|
Die MDR gilt direkt, ohne dass diese, wie z. B. die MDD, zuerst in nationales Gesetz (MPG) überführt wird.
|
|
6
|
Ab wann ist die MDR verpflichtend anzuwenden?
|
Die MDR ist nach einer vierjährigen Übergangszeit ab dem 26. Mai 2021 (Geltungsbeginn) verpflichtend anzuwenden.
Ausnahmen bestehen für Medizinprodukte der Klassen IIa bis III (z. B. Biatain Ag), für die vor dem MDR-Geltungsbeginn ein MDD-Zertifikat ausgestellt wurde, das bis maximal drei Jahre nach dem Geltungsbeginn der MDR weitergilt.
|
|
7
|
Was ist für Händler neu?
|
Die MDR definiert erstmalig spezifische Anforderungen für Händler.
|
|
8
|
Wie ist "Händler" nach der MDR definiert?
|
„Händler“ bezeichnet jede natürliche oder juristische Person in der Lieferkette, die ein Produkt bis zum Zeitpunkt der Inbetriebnahme auf dem Markt bereitstellt, mit Ausnahme des Herstellers oder des Importeurs;
Dies sind also Unternehmen wie z. B. Sanitätshändler, Homecare-Unternehmen, Großhändler und Apotheken.
|
|
9
|
Was ist nicht unter "Händler" zu verstehen?
|
Unternehmen, die die Produkte nicht kaufen, besitzen oder verkaufen, sondern nur im Auftrag eines anderen Unternehmens handeln, werden nicht als Händler betrachtet. Dies beinhaltet Transport und gemietete Lagermöglichkeiten.
|
|
10
|
Was sind die Pflichten eines Händlers?
|
- Hersteller, Importeure oder Bevollmächtigte über etwaige Reklamationen und Verstöße (gegen die MDR) der Produkte zu informieren
- die zuständige Behörde über ernsthafte Risiken mit Produkten zu informieren
- Überprüfung der Konformität der abzugebenden Medizinprodukte (kann eine Stichprobe sein - keine 100%-ige Kontrolle)
- dafür zu sorgen, dass die Lagerungs- und Transportbedingungen den Vorgaben des Herstellers entsprechen
- die Rückverfolgbarkeit (Lieferant / Kunde) aller abgegebenen Medizinprodukte sicherzustellen
- Registrierung des Händlers, falls es von den nationalen Behörden verlangt wird.
|
|
11
|
Welche Informationen stellt Coloplast in Zusammenhang mit Artikel 14 MDR zur Verfügung?
|
Unsere Handelspartner können u. a. folgende Materialien anfordern bzw. herunterladen:
- Gebrauchsanweisungen: Download hier
- Coloplast-Konformitätserklärungen: Download hier
- UDI und eine „dem Hersteller Coloplast und dem Produkt eigene UDI-Produktkennung“ anfordern.
|
|
12
|
Was bedeutet UDI und EUDAMED?
|
Unique Device Identification (UDI) Produktidentifizierungsnummer ist ein weltweites System für eine einheitliche Produktkennzeichnung für Medizinprodukte. Die UDI soll sowohl mit maschinenlesbaren Kennzeichen (beispielsweise Barcode) als auch in Klarschrift auf dem Produkt aufgebracht werden. Die UDI dient als Schlüssel zu einer UDI-Datenbank (EUDAMED), die eine Vielzahl von Informationen zu den Produkten enthalten wird.
Die europäische Datenbank für Medizinprodukte, kurz EUDAMED, ist die zentrale Plattform für alle Informationen über Hersteller, Produkte, Bescheinigungen und klinische Prüfungen.
|
|
13
|
Welche UDI Fristen sind zu beachten?
|
Die Frist für die Meldung von Daten an EUDAMED wurde verschoben, da sich die Entwicklung von EUDAMED verzögert. Die Kennzeichnungs-Fristen für die UDI-Implementierung auf Etiketten bleiben unverändert, die Datenübermittlung wurde jedoch um zwei Jahre verschoben. Die neuen Daten sind:
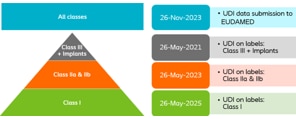
Coloplast hat sich dazu entschieden, alle UDI-Änderungen der Kennzeichnung gleichzeitig vorzunehmen. Dies beinhaltet das Platzieren eines UDI-Barcodes auf den Etiketten. Die Verifizierung der UDI-Barcodes durch EUDAMED erfolgt jedoch gemäß den oben genannten Fristen.
|
|
14
|
Was bedeutet "2 D Barcode"?
|
Ein Barcode ist ein aus Strichen bestehender optischer Datenträger (auch Strichcode genannt). 2-dimensionale Matrixcodes werden u.a. auch als 2D Barcodes bezeichnet. Dazu zählt auch der Data Matrix Code.
Beispiel:

|
|
15
|
Welche Produkte sind von der MDR betroffen und welche Termine sind für die jeweiligen Produkt-/Risikoklassen wichtig?
|
Medizinprodukte werden in vier Klassen eingeteilt: I, IIa, IIb und III, wobei Klasse I noch weitere Untergruppen Is, Im und Ir enthält.
Die Einteilung richtet sich nach verschiedenen Kriterien, wie beispielsweise der Anwendungsdauer, dem Grad der Invasivität oder der Wiederverwendbarkeit.
Die meisten Klassen (Is, Im, Ir, IIa, IIb, III) benötigen eine Bewertung und Zertifizierung durch eine akkreditierte „Benannte Stelle (Notified Body)". Coloplast arbeitet mit „Presafe“ zusammen.
Allerdings gelten alle ausgestellten Zertifikate (Is, Im, Ir, IIa, IIb, III) nach der bisherigen MDD (Medical Device Directive) gemäß ihrer angegebenen Laufzeit weiter, bis spätestens 2024. Dies gilt allerdings nur dann, wenn am Produkt keine Änderungen vorgenommen werden. Ist dies der Fall, muss zwingend eine Zertifizierung nach MDR erfolgen.
Anders sieht es für die Klasse I, also unsterile Produkte der Klasse I, aus. Diese Produkte müssen bis zum Stichtag am 26. Mai 2021 MDR-konform sein. Die Konformität belegen die Hersteller durch eine Konformitätserklärung („Declaration of conformity“) bzw. per Selbst-Zertifizierung und CE-Kennzeichnung der Produkte. Beispiele wie folgt:
|